
バクテリアStaphylococcusaureusからのタンパク質
11月下旬、GoogleのDeepMindチーム は、AlphaFoldディープラーニングシステムが、計算生化学の難しい問題であるタンパク質フォールディング問題の解決において前例のないレベルの精度を達成したことを発表しました 。
問題は何ですか、そしてなぜそれを解決するのがとても難しいのですか?
タンパク質はアミノ酸の長鎖です。あなたのDNAはこれらの配列をコードし、RNAはこの遺伝的青写真に従ってタンパク質を作るのを助けます。タンパク質は線形鎖の形で合成されますが、その後、複雑な球状構造に折りたたまれます(記事の冒頭の写真を参照)。
チェーンの一部が丸まってタイトなスパイラルになることがあります。」 α-ヘリックス。「他の部分は前後に曲がって、広くて平らな形を形成することができます。」 β-シート":
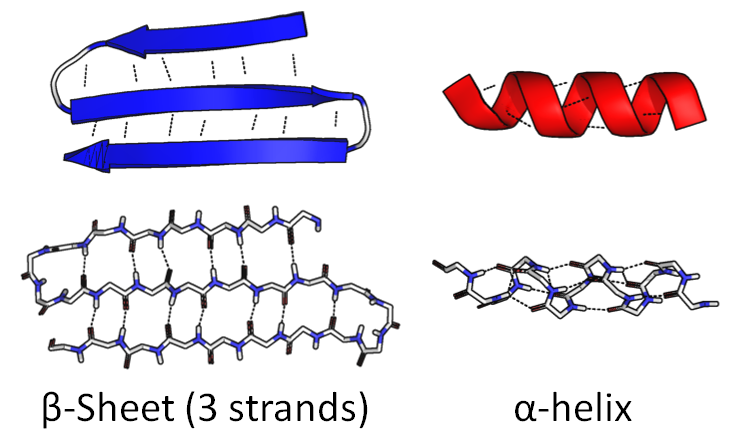
アミノ酸配列自体を一次構造と呼び ます。これらの図は二次構造と呼ばれ ます。
これらのコンポーネント自体も折りたたまれて、独特の複雑な形状を形成します。これは三次構造と呼ばれ ます:
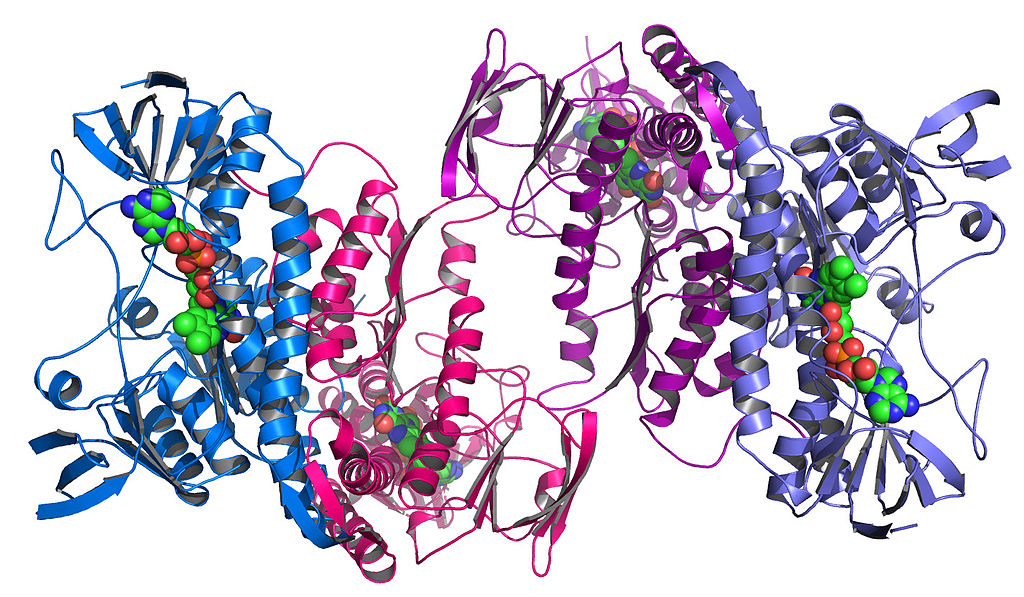
細菌Colwellia psychrerythraeaRRM3

タンパク質 から取られた酵素
乱雑に見えます。なぜこの絡み合ったアミノ酸の球がそれほど重要なのですか?
タンパク質の構造はランダムではありません!各タンパク質は、明確で、独特で、大部分が予測可能な構造に折りたたまれます。これは、適切に機能するために不可欠です。その物理的形態のために、タンパク質はそれが結合できる構造によく適しています。他の物理的特性、特にタンパク質全体の電荷の分布も重要です。写真では、正電荷は青、負電荷は赤で示されています。

米の植物1の脂質担体タンパク質の表面電荷分布
タンパク質が本質的に自己組織化ナノマシンである場合、アミノ酸配列の主な目的は、その独特の形状、電荷分布、およびタンパク質の機能を決定するその他すべてを生成することです。このプロセスがどのように正確に行われるかはまだ完全には明らかではありません-今日、それは研究の活発な分野です。
いずれにせよ、構造を理解することは、それがどのように機能するかを理解するために重要です。ただし、DNAシーケンスは、タンパク質の一次構造のみを定義します。その二次および三次構造、つまり、このもつれがとる正確な形状をどのようにして知ることができますか?
この問題はタンパク質フォールディング問題と呼ばれ、測定と予測という2つの基本的なアプローチがあります。
実験的方法は、タンパク質の構造を測定することができます。ただし、これを行うのはそれほど簡単ではありません。構造は光学顕微鏡では見えません。長い間、X線結晶学は構造を研究するための主な方法でした。それに加えて、核磁気共鳴が使用され、最近、新しい技術、低温電子顕微鏡法が登場しました 。

SARSプロテアーゼのX線回折パターン
しかし、これらの方法は、費用がかかり、複雑で、時間がかかり、さらに、すべてのタンパク質で機能するわけではありません。特に、細胞膜に埋め込まれたタンパク質(COVID-19ウイルスが結合するのと同じアンギオテンシン変換酵素2(ACE2)受容体 )は、脂質二重層に折りたたまれます 細胞、そして結晶化することは非常に困難です。

細胞膜の構造
したがって、シーケンスされたタンパク質のごく一部の構造を分解することができました 。 ユニバーサルタンパク質データベースは一方で、1.8億配列を含む三次元タンパク質構造のデータベースが含まれている だけ17万位置を。
より良い方法が必要です。
* * *
タンパク質の二次および三次構造は、基本的に、シーケンシングを通じて私たちに知られている一次構造の関数であることを思い出してください。タンパク質の構造を測定する代わりに、それを予測できるとしたらどうでしょうか。
これは、タンパク質の構造を予測するタスクです。計算生化学者は何十年もの間それに取り組んできました。
どのようにアプローチできますか?
明らかな方法は、プロセスの物理を直接シミュレートすることです。位置、電荷、化学結合を考慮して、各原子の力をシミュレートします。加速と速度を数え、システムの進化を段階的にスクロールします。これは「分子ダイナミクス」と呼ばれます。 DE ShawResearchによる

スーパーコンピューター「Anton」

スーパーコンピューター
IBMBlue GeneOnlineパズルFoldit
問題は、このアプローチが非常に計算集約的であるということです。典型的なタンパク質には、数百のアミノ酸、つまり数千の原子が含まれています。環境も重要です。折りたたむと、タンパク質は周囲の水と相互作用します。したがって、約3万個の原子の挙動をシミュレートする必要があります。この場合、静電相互作用が原子の各ペア間で発生します。つまり、概算では、4億5000万ペアが得られます。これは、複雑さO(N2)の問題です。その複雑さをO(N log N)に減らす巧妙なアルゴリズムがあります。加えて、10の9 -10 12のステップは、シミュレーションのために計算されなければなりません 。並外れた頭痛。
わかりましたが、折り畳みプロセス全体をシミュレートする必要はありません。別のアプローチは、潜在的なエネルギーが最小の構造を見つけることを提案します。通常、オブジェクトは最小のエネルギーで静止する傾向があるため、このヒューリスティックなアプローチは正当化されます。エネルギーは同じ分子動力学モデルで計算でき、相互作用の大きさがわかります。このアプローチでは、多数の候補を試して、エネルギーが最も少ない構造を選択できます。もちろん、問題は構造をどこから取得するかです。それらの数が多すぎるだけです-分子生物学者のCyrusLevintolは、約10,300個あると計算してい ます。当然、ランダムなブルートフォースよりも賢いアプローチを使用できます。しかし、まだそれらの数が多すぎます。
したがって、そのような計算を高速化するためにすでに多くの試みがなされてきました。 DE Shaw ResearchのスーパーコンピューターであるAntonは、特別なハードウェア、つまり特別な集積回路を使用しています。 IBMはBlueGeneバイオスーパーコンピューターも使用しています。スタンフォードは、家庭用コンピューターの分散型電源を使用して、Folding @Homeプロジェクトを立ち上げました。 UWのFolditプロジェクトは、フォールディングをゲームに変えて、計算に人間の直感を追加しました。
しかし、長い間、広範囲のタンパク質構造を高精度で予測できる技術はありませんでした。年に2回開催されるCASPコンペティションでは、アルゴリズムの結果が実験的に測定された構造と比較され、1位は30〜40%の精度で予測を受けました。最近まで:

無料モデリングカテゴリで最高のチーム中央値予測精度
。AlphaFoldはどのように機能しますか?複数のディープニューラルネットワークを使用して、各タンパク質に関連するさまざまな機能を学習します。重要な機能の1つは、アミノ酸のペア間の結果の距離を予測することです。これにより、アルゴリズムが最終的な構造になります。アルゴリズムの1つの変形(ジャーナル Nature and Proteinsに記載)では、この予測の潜在的な関数が導き出され、最も単純な勾配降下が適用され、驚くほどうまく機能しました。
以前の方法に対するAlphaFoldの主な利点は、構造について仮定する必要がないことです。いくつかの方法は、タンパク質をセクションに分割し、それぞれを数え、次にすべてを元に戻すことによって機能します。 AlphaFoldはこれを必要としません。
どうやら、DeepMindは折り畳みの問題が解決されたと考えているようです。これは私には単純化しすぎているように見えますが、いずれにせよ、その進歩は重要です。 Googleと提携していない専門家は、「素晴らしい」や「革命的な」などの 表現を使用します 。
遺伝子工学には、CRISPRとタンパク質フォールディングという2つの強力なツールがあります。おそらく2020年代は、1970年代がコンピューティングのためだったのと同じようにバイオテクノロジーのためになるでしょう。
このブレークスルーについてDeepMindの研究者の皆さん、おめでとうございます!